COMBINE Metagenomics Workshop, 2024
University of Queensland, Queensland, Australia, 2024
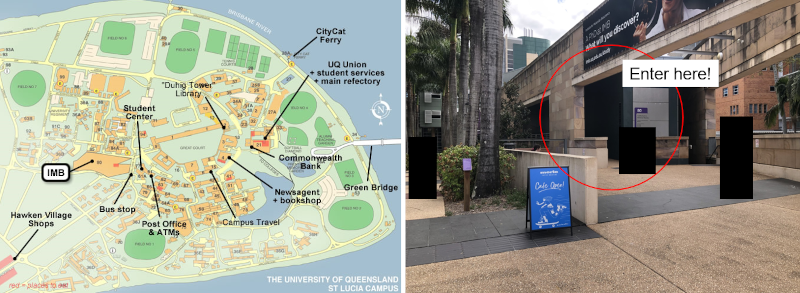
Workshop Location
Room 3.142 Institute for Molecular Bioscience, University of Queensland, St Lucia, QLD.
You can find the room here:
Workshop Schedule
Please note: The workshop schedule is subject to change depending on how quickly or slowly we progress.
Day 1: Monday September 9th
| Time | Topic |
|---|---|
| 0900-0915 | Welcome and Introductions. |
| 0915-1000 | Introduction to metagenomics, Linux, and Bash |
| 1000-1030 | Bash hands on and practice |
| 1030-1100 | Morning coffee |
| 1100-1200 | Introduction to methods for identifying species GTDB, SingleM |
| 1200-1330 | Lunch |
| 1330-1400 | Hands-on with SingleM |
| 1400-1500 | Introduction to binning |
| 1500-1530 | Afternoon tea |
| 1530-1600 | Microbial binning |
| 1600-1700 | Invited Presentation: Professor Elizabeth Dinsdale |
Day 2: Tuesday September 10th
| Time | Topic |
|---|---|
| 0900-0930 | Recap of Day 1 |
| 0930-1030 | Removing the human genome |
| 1030-1100 | Morning coffee |
| 1100-1130 | Introduction to methods for functional analysis |
| 1130-1200 | SUPER-FOCUS hands-on |
| 1200-1300 | Lunch |
| 1300-1400 | Viral identification using Hecatomb |
| 1400-1500 | Hecatomb hands-on |
| 1500-1530 | Afternoon tea |
| 1530-1645 | Hands-on data visualisation |
| 1645-1700 | Wrap up and summary |
Download
If you are using a MS Windows machine, please download and install MobaXterm before we start. If you are using a Mac, you are good to go!
Metagenomics
We are going to jump right in with metagenomics, but here is a brief introduction if you want to read something while Rob is talking.
We have created servers for you with all the software and data that you will need for these excercises.
There are two machines that you can use, if you don’t have access to a server:
IP Addresses:
1:
2:
3:
4:
5:
6:
7:
Learning BASH
If you need more helo with bash, you can follow the Pony!
We have installed PonyLinux. You only need two commands to get started.
First, log into the accounts provided for you and then follow the commands:
Next type cd PonyLinux and press enter (or on some computers it is called return).
Now, type ./Ponylinux.sh and press enter (or return).
Installing software
Our first excercise is installing software using mamba.
Install conda, fastp, minimap2, samtools using conda
We will use all of these programs today.
SingleM
take a look at the manual for detailed singlem instructions.
Install singleM with mamba:
mamba create -n singlem -c bioconda singlem
mamba activate singlem
Before you use it, make sure you add this command:
export SINGLEM_METAPACKAGE_PATH=/storage/data/metapackage
Now run singleM on the CF data:
singlem pipe -1 /storage/data/cf_data/CF_Data_R1.fastq.gz -2 /storage/data/cf_data/CF_Data_R2.fastq.gz -p output_profile.tsv
BinChicken
Here are the detailed instructions for BinChicken
Downloading Data
All the data we are going to use in the workshop is present on the servers in /storage/data/cf_data
Use fastp to trim bad sequences and remove the adapters.
We are going to use the Illumina Adapters, and trim out:
- Sequences that are less 100 bp
- Sequences that contain 1 N
- Trim the adapters off the 3’ and 5’ ends of the sequences
Do you remember how to download the Illumina Adapters? The URL is https://github.com/linsalrob/ComputationalGenomicsManual/raw/master/SequenceQC/IlluminaAdapters.fa
Once you have downloaded the adapters, we can use this command:
mkdir fastp
fastp -n 1 -l 100 -i /storage/data/cf_data/reads/788707_20180129_S_R1.fastq.gz -I /storage/data/cf_data/reads/788707_20180129_S_R2.fastq.gz -o fastp/788707_20180129_S_R1.fastq.gz -O fastp/788707_20180129_S_R2.fastq.gz --adapter_fasta IlluminaAdapters.fa
When fastp runs, you will get an HTML output file called fastp.html. This shows some statistics about the run.
Filter out the human and non-human sequences.
The NCBI has several human genome versions specifically designed for inclusion in pipelines like this.
A. GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz
A gzipped file that contains FASTA format sequences for the following:
1. chromosomes from the GRCh38 Primary Assembly unit.
Note: the two PAR regions on chrY have been hard-masked with Ns.
The chromosome Y sequence provided therefore has the same
coordinates as the GenBank sequence but it is not identical to the
GenBank sequence. Similarly, duplicate copies of centromeric arrays
and WGS on chromosomes 5, 14, 19, 21 & 22 have been hard-masked
with Ns (locations of the unmasked copies are given below).
2. mitochondrial genome from the GRCh38 non-nuclear assembly unit.
3. unlocalized scaffolds from the GRCh38 Primary Assembly unit.
4. unplaced scaffolds from the GRCh38 Primary Assembly unit.
5. Epstein-Barr virus (EBV) sequence
Note: The EBV sequence is not part of the genome assembly but is
included in the analysis set as a sink for alignment of reads that
are often present in sequencing samples.
B. GCA_000001405.15_GRCh38_full_analysis_set.fna.gz
A gzipped file that contains all the same FASTA formatted sequences as
GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz, plus:
6. alt-scaffolds from the GRCh38 ALT_REF_LOCI_* assembly units.
C. GCA_000001405.15_GRCh38_full_plus_hs38d1_analysis_set.fna.gz
A gzipped file that contains all the same FASTA formatted sequences as
GCA_000001405.15_GRCh38_full_analysis_set.fna.gz, plus:
7. human decoy sequences from hs38d1 (GCA_000786075.2)
D. GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna.gz
A gzipped file that contains all the same FASTA formatted sequences as
GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz, plus:
7. human decoy sequences from hs38d1 (GCA_000786075.2)
For this work, we are going to use GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna.gz which contains everything.
We have made this data available for you in /storage/data/human
Install minimap and samtools
This also installs the conda channels for you in the right order!
conda config --add channels bioconda
conda config --add channels conda-forge
mamba create -n minimap2 minimap2 samtools
Use minimap2 and samtools to filter the human sequences
minimap2 --split-prefix=tmp$$ -a -xsr /storage/data/human/GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set.fna.gz fastp/788707_20180129_S_R1.fastq.gz fastp/788707_20180129_S_R2.fastq.gz | samtools view -bh | samtools sort -o 788707_20180129.bam
samtools index 788707_20180129.bam
Here is the samtools specification, and the description of the columns is on page 6.
Now, we use samtools flags to filter out the human and not human sequences. You can find out what the flags mean using the samtools flag explainer
human only sequences
mkdir human not_human
samtools fastq -F 3588 -f 65 788707_20180129.bam | gzip -c > human/788707_20180129_S_R1.fastq.gz
echo "R2 matching human genome:"
samtools fastq -F 3588 -f 129 788707_20180129.bam | gzip -c > human/788707_20180129_S_R2.fastq.gz
sequences that are not human
samtools fastq -F 3584 -f 77 788707_20180129.bam | gzip -c > not_human/788707_20180129_S_R1.fastq.gz
samtools fastq -F 3584 -f 141 788707_20180129.bam | gzip -c > not_human/788707_20180129_S_R2.fastq.gz
samtools fastq -f 4 -F 1 788707_20180129.bam | gzip -c > not_human/788707_20180129_S_Singletons.fastq.gz
Using snakemake
In the above example, we started with two fastq files, removed adapter sequences, mapped them to the human genome, and then separated out the human and not human sequences.
We can combine all of that into a single snakemake file, and it will do all of the steps for us.
See the Snakemake section for details on how to run these two commands in a single pipeline.
Super Focus
Install superfocus using mamba:
mamba create -n superfocus super-focus
mamba activate superfocus
And now run it:
Note: super-focus requires uncompressed gzip files at the moment.
superfocus -q not_human/ -dir superfocus -a mmseqs -t 16 -db DB_95 -b /storage/data/superfocus/
Hecatomb
We have already run the hecatomb pipeline for you, and the data is in /storage/data/hecatomb, but you need to download the data to your computer before we can analyse the files.
There are three files:
and you can copy them like this:
cp -r /storage/data/hecatomb/ .
Note: don’t forget the last dot
Start a Google Colab Notebook
Connect to Google Colab
Load the data into pandas dataframes:
import os
import sys
import matplotlib.pyplot as plt
import scipy.stats as stats
import statsmodels.stats.api as sms
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
Copy the files into your Jupyter notebooks and then find the files
os.chdir('Data')
os.listdir()
Read the data into pandas data frames
data = pd.read_csv('bigtable.tsv.gz',compression='gzip',header=0,sep='\t')
# without downloading - copy link address from above
# data = pd.read_csv('https://github.com/linsalrob/CF_Data_Analysis/raw/main/hecatomb/bigtable.tsv.gz?download=',compression='gzip',header=0,sep='\t')
metadata = pd.read_csv('CF_Metadata_Table-2023-03-23.tsv.gz',compression='gzip',header=0,sep='\t')
vmr = pd.read_csv('VMR_MSL39_v1.ascii.tsv.gz', compression='gzip',header=0,sep='\t')
Take some time to look at the tables.
How many columns are there in each? How many rows? What are the data types in the rows and columns?
First, filter the bigtable for just the Viruses
viruses = data[(data.kingdom == "Viruses")]
Group them by alignment type, length, and family
virusesGroup = viruses.groupby(by=['family','alnType','alnlen','pident'], as_index=False).count()
Now use seaborn to make some pretty plots!
First, we start by creating a style that we like.
#styling
sizeScatter = 10 * virusesGroup['count']
sns.set_style("darkgrid")
sns.set_palette("colorblind")
sns.set(rc={'figure.figsize':(12,8)})
# and now plot the data
g = sns.FacetGrid(virusesGroup, col="family", col_wrap=10)
g.map_dataframe(sns.scatterplot, "alnlen", "pident", alpha=.1, hue="alnType", sizes=(100,500), size=sizeScatter)
plt.legend(bbox_to_anchor=(5.0,1), loc=0, borderaxespad=2,ncol=6, shadow=True, labelspacing=1.5, borderpad=1.5)
plt.show()
That’s a lot of data!
Restrict this plot to just E value < 1 x 10-20
virusesFiltered = viruses[viruses.evalue<1e-20]
virusesGroup = virusesFiltered.groupby(by=['family','alnType','alnlen','pident'], as_index=False).count()
g = sns.FacetGrid(virusesGroup, col="family", col_wrap=10)
g.map_dataframe(sns.scatterplot, "alnlen", "pident", alpha=.1, hue="alnType", sizes=(100,500), size=sizeScatter)
for ax in g.axes.flat:
ax.tick_params(axis='both', labelleft=True, labelbottom=True)
ax.axhline(y=75, c='red', linestyle='dashed', label="_horizontal")
ax.axvline(x=150, c='red', linestyle='dashed', label="_vertical")
plt.legend(bbox_to_anchor=(5.0,1), loc=0, borderaxespad=2,ncol=6, shadow=True, labelspacing=1.5, borderpad=1.5)
plt.show()
More detailed analysis
There are two proteins that are bogus. We don’t know why, so we just ignore. For the next steps, we also only look at the amino acid hits, not the nucleotide hits.
data = pd.read_csv('bigtable.tsv.gz',compression='gzip',header=0,sep='\t')
blacklist = ['A0A097ZRK1', 'G0W2I5']
virusesFiltered = data[(data.alnType == "aa") & (data.kingdom == "Viruses") & (~data.targetID.isin(blacklist) & (data.evalue < 1e-20))]
Patient and Sample Date
Now we create new columns for the patient and the sample date
virusesFiltered[['patient', 'date', 'Sputum or BAL']] = virusesFiltered['sampleID'].str.split('_', expand=True)
Merge the real data and the host data
virusesFiltHost = pd.merge(virusesFiltered, vmr[['Family', 'Host source']], left_on="family", right_on="Family", how='left')
Take a look at this new table. What have we accomplished? What was done? How did it work?
Correct more errors
There are two families that are incorrectly annotated in this table. Let’s just manually correct them:
really_bacterial = ['unclassified Caudoviricetes family', 'unclassified Crassvirales family']
virusesFiltHost.loc[(virusesFiltHost['family'].isin(really_bacterial)), 'Host source'] = 'bacteria'
Group-wise data
Now lets look at some data, and put that into a group. We’ll also remember what we’ve looked
to_remove = []
bacterial_viruses = virusesFiltHost[(virusesFiltHost['Host source'] == 'bacteria')]
to_remove.append('bacteria')
Can you make a plot of just the bacterial viruses like we did?
How many reads map per group, and what do we know about them
host_source = "archaea"
rds = virusesFiltHost[(virusesFiltHost['Host source'] == host_source)].shape[0]
sps = virusesFiltHost[(virusesFiltHost['Host source'] == host_source)].species.unique()
fams = virusesFiltHost[(virusesFiltHost['Host source'] == host_source)].family.unique()
print(f"There are {rds} reads that map to {host_source} viruses, and they belong to {len(sps)} species ", end="")
if len(sps) < 5:
spsstr = "; ".join(sps)
print(f"({spsstr}) ", end="")
print(f"and {len(fams)} families: {fams}")
to_remove.append(host_source)
Can you repeat this for each of (plants OR plants (S)), algae, protists, invertebrates, fungi,
Filter out things we don’t want
Once we remove the above, what’s left?
virusesFiltHost = virusesFiltHost[~virusesFiltHost['Host source'].isin(to_remove)]
virusesFiltHost['Host source'].unique()
Now look at what we have
#filter
virusesGroup = virusesFiltHost.groupby(by=['family','alnlen','pident'], as_index=False).count()
#styling
sizeScatter = 10 * virusesGroup['count']
sns.set_style("darkgrid")
sns.set_palette("colorblind")
sns.set(rc={'figure.figsize':(20,10)})
g = sns.FacetGrid(virusesGroup, col="family", col_wrap=6)
g.map_dataframe(sns.scatterplot, "alnlen", "pident", alpha=.8, sizes=(100,500), size=sizeScatter)
for ax in g.axes.flat:
ax.tick_params(axis='both', labelleft=True, labelbottom=True)
ax.axhline(y=80, c='red', linestyle='dashed', label="_horizontal")
ax.axvline(x=150, c='red', linestyle='dashed', label="_vertical")
g.fig.subplots_adjust(hspace=0.4)
g.set_axis_labels("Alignment length", "Percent Identity")
plt.legend(bbox_to_anchor=(5.0,1), loc=0, borderaxespad=2,ncol=6, shadow=True, labelspacing=1.5, borderpad=1.5)
plt.show()
Now look at each family separately
virusesFiltHost[virusesFiltHost.family == 'Retroviridae']